Large Language Models Meet Virtual Cell: A Survey
-
ArXiv URL: http://arxiv.org/abs/2510.07706v1
-
作者: Yuanjie Zou; Zheng Hui; Guang Yang; Zhonghao Zhan; Weiye Bao
-
发布机构: Imperial College London; King’s College London; New Jersey Institute of Technology; Royal Brompton Hospital; University College London; University of Cambridge
TL;DR
本文全面综述了大型语言模型(LLM)在构建“虚拟细胞”(virtual cell)——一个能够表示、预测和推理细胞状态与行为的计算系统——中的应用,并提出了一个将现有方法分为“作为预言机的LLM”(LLMs as Oracles)和“作为智能体的LLM”(LLMs as Agents)两大范式的统一分类体系。
引言
细胞是生命的基本单位,理解其复杂的分子程序是生物学的核心目标。然而,细胞系统的高维性和复杂性使这一任务极具挑战性。人工智能(AI),特别是大型语言模型(LLM)的进步,为构建“虚拟细胞”提供了前所未有的机遇。虚拟细胞是一种在硅基(in silico)中模拟细胞结构、功能和动态的计算系统,它有望加速药物发现和实现个性化医疗。
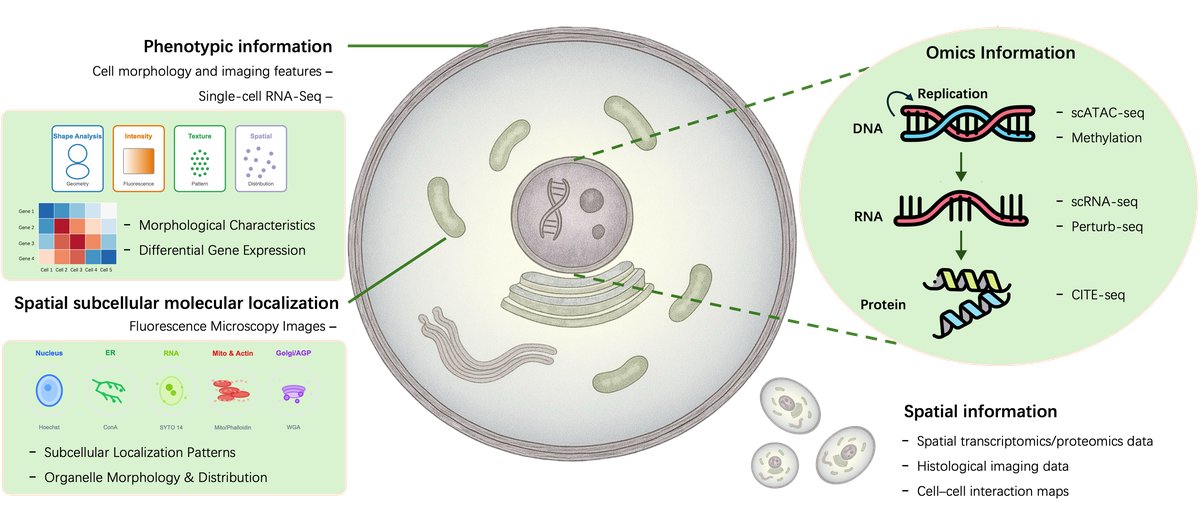
虚拟细胞的概念并非全新,早期的系统生物学尝试通过机理或统计模型重构细胞行为,但受限于不完整的知识和稀疏的数据。随着组学(omics)数据和LLMs的爆发式增长,研究者可以直接在海量生物语料库上训练基础模型,使虚拟细胞演变为一个数据驱动、生成式和具备推理能力的框架。
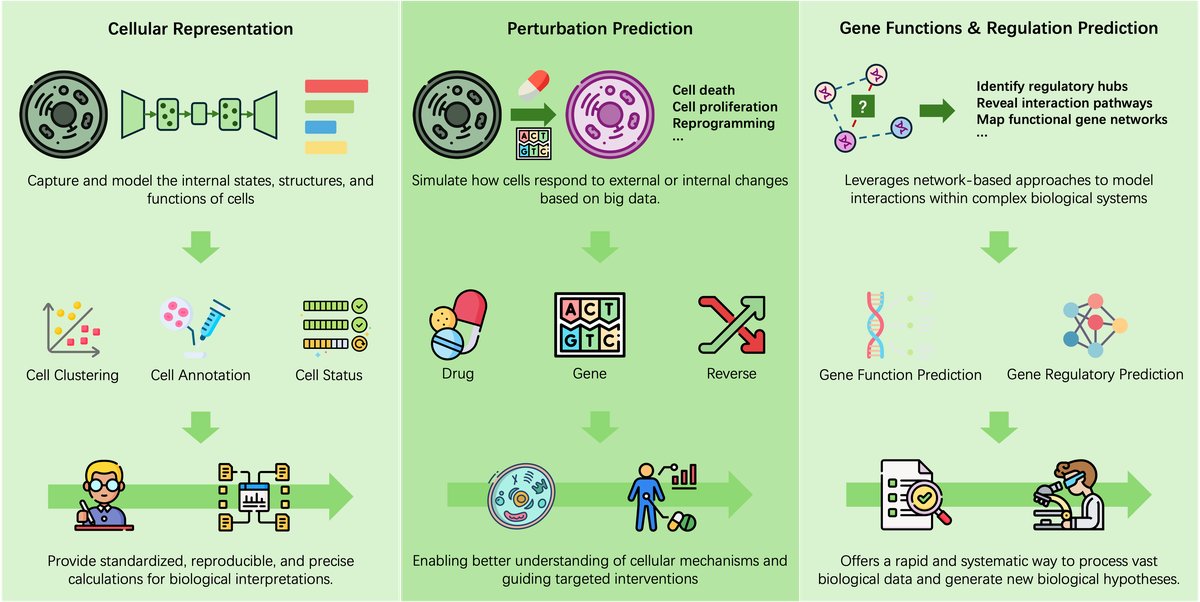
JUMP-Cell Painting和Chan Zuckerberg Initiative (CZI)等大型研究项目提供了丰富的标准化数据集,如CELLxGENE和Tabula Sapiens,极大地推动了这一领域的发展。这些进展共同为精确模拟细胞系统奠定了基础。其中,三大核心任务构成了AI驱动的虚拟细胞的操作支柱:
- 细胞表示(Cellular Representation):准确进行细胞注释、分类和状态预测。
- 扰动预测(Perturbation Prediction):模拟基因或药物干预的效果,支持因果推断和疗法发现。
- 基因调控推断(Gene Regulation Inference):破译基因作用,重建调控网络,揭示细胞过程的内在机制逻辑。
本文的主要贡献如下:
- 首次系统总结了LLM和智能体如何变革虚拟细胞的开发,连接了人工智能与细胞生物学。
- 提出了一个将现有方法分为两大范式的分类体系:作为预言机的LLM用于模拟细胞状态和分子相互作用,作为智能体的LLM用于自主推理、规划和实验。
- 整合了当前进展,并指出了在可扩展性、可解释性和生物保真度方面的开放性挑战,为下一代AI驱动的虚拟细胞系统提供了战略见解和发展路线图。
作为预言机的LLM方法
在此范式中,LLM被视为虚拟细胞的计算“预言机”(Oracle),直接对细胞系统的内部状态和动态进行建模。它们处理DNA、RNA或单细胞转录组等生物序列,模型本身作为预测引擎,从原始数据中学习细胞组分和相互作用的表示,而不依赖外部工具。
核苷酸序列
DNA:作为细胞的蓝图,其长程依赖关系(如增强子在100kb外调控基因)是建模的关键挑战。早期模型如DeepSEA使用卷积网络(CNN),而后续模型如Enformer结合了CNN和Transformer,将输入序列扩展到200kb。最近,纯Transformer编码器模型(如DNABERT系列和Nucleotide Transformer, NT)通过掩码语言建模(Masked Language Modeling, MLM)进行预训练,其中NT模型参数量达到25亿。HyenaDNA则采用新的Hyena算子和自回归的下一Token预测(next-token prediction, NTP),能够处理长达100万个Token的序列。
RNA:RNA在细胞中功能多样。基于Transformer编码器的RNABERT模型在2370万个非编码RNA序列上训练。Riboformer则扩展至6.5亿参数。U-RNA通过在预训练中加入基序感知的MLM来增强对功能性RNA元件的敏感性。
蛋白质-蛋白质相互作用(PPI)
蛋白质-蛋白质相互作用(Protein-protein interactions, PPIs)是细胞信号传导和代谢途径的基础。
- 基于多序列比对(MSA)的方法:AlphaFold-Multimer利用MSA预测蛋白质复合物的3D结构,其pDockQ指标可用于可靠区分PPI。
- 基于蛋白质语言模型(PLM)的方法:为解决MSA计算成本高和对无同源序列不准确的问题,D-SCRIPT等模型被开发出来。ConPlex利用MLM框架预测跨物种的PPI和基因重要性。
多领域分子
整合DNA、RNA和蛋白质的联合表示是捕获细胞复杂动态的关键。GENA-LM及其后续版本Evo通过NTP方法在万亿级核苷酸序列上训练,学习跨生命领域分子的联合表示。同时,AlphaFold 3、RoseTTAFold All-Atom等模型已能预测所有类型生物分子及其相互作用的结构。
单组学
组学数据,尤其是单细胞RNA测序(scRNA-seq),是细胞建模基础LLM的主要数据源。单细胞组学数据通常表示为一个细胞-基因表达矩阵 $\mathbf{X}\in\mathbb{R}^{N\times G}$,其中 $N$ 是细胞数,$G$ 是基因数。
面对组学数据的噪声和批次效应等挑战,scBERT和scGPT采用了类似掩码自编码器(Masked Autoencoder, MAE)的架构。Geneformer则将训练规模扩展至3000万个细胞,而scGPT-650M进一步扩展至5000万细胞和6.5亿参数。在架构创新方面,scETM采用了改进的ERetNet骨干。
此外,将生物先验知识融入模型也证明了其有效性。例如,scELMo和scBERT-Protein通过整合蛋白质语言模型(PLM)的嵌入来增强跨物种泛化能力。pert-GPT则专为扰动响应预测而设计。在表观基因组学领域,EpiGeNet集成序列、染色质和基因组信息,实现了对跨细胞类型表观基因组状态的上下文感知预测。
多组学
单一组学无法完全捕获细胞状态,因此多组学整合至关重要。
- 数据整合:scGPT采用GPT风格的自回归架构,将不同组学数据tokenize到共享词汇表中进行统一建模。scGraph则集成了图神经网络(GNN)来显式建模空间转录组数据中的细胞邻里关系。U-CROMU模型则扩展至9种不同的单细胞组学,能够对下游任务进行零样本预测。
- 多组学翻译:旨在从可用数据中推断或重建缺失的组学模态。例如,scTranslator利用Transformer解码器将scRNA-seq输入翻译为相应的蛋白质组谱。
多模态
将科学文本作为一种额外模态,可以增强模型的泛化能力和生物学基础。
- 文本与细胞数据对齐:scCLIP采用对比学习框架对齐scRNA-seq谱和文本描述的潜在表示。Cellar则通过数值分箱将基因表达值映射到固定词汇表,从而能够直接微调GPT-2。scBLIP和CellLM则通过交叉注意力机制连接单细胞LLM和通用文本LLM,实现细胞与文本模态间的双向翻译。
- 科学推理:为了赋予模型更强的自主发现能力,一些工作开始引入强化学习(Reinforcement Learning, RL)和思维链(Chain-of-Thought, CoT)提示。例如,scGRPO利用RL对齐scRNA-seq表示与自然语言理解能力。其他模型通过蒸馏由大型LLM生成的CoT推理过程,赋予小模型推理能力。
作为智能体的LLM方法
在此范式中,LLM作为虚拟细胞的智能“智能体”(Agent),通过协调外部工具、数据库和模拟环境来完成超越传统建模和预测的复杂科研任务。与被动生成输出的基础模型不同,LLM智能体在一个自适应、目标驱动的框架内主动规划、推理和行动。

架构
- 单智能体系统:单个LLM作为统一智能体,通过内部推理或动态提示管理整个工作流程。
- 多智能体系统:将职责分配给多个专门的LLM(如规划器、分析器、执行器),它们通过对话或共享内存进行协作,提高了可扩展性和透明度。
文献与知识
为确保生物学有效性,LLM智能体与科学文献和结构化数据库接口。检索增强生成(Retrieval-Augmented Generation, RAG)是常用策略,它通过在推理时检索相关信息来提高回答的事实准确性。例如,BioChat索引了超过2200万篇科学文章,而PubMed-GPT则提供对PubMed等知识库的交互式访问。scHarvester智能体则能自主收集和处理scRNA-seq数据,构建一个持续扩展的数据库。
实验设计
LLM智能体可将高层次的生物学问题转化为可操作的实验计划。
- 假设生成:SPA-GPT能够解释空间转录组学数据,提出关于组织结构和细胞相互作用的新机制假设。
- 流程指导:GeneTAC可为CRISPR基因编辑工作流自动分解设计过程。Perturb-Assistant能够通过规划迭代式的Perturb-Seq实验来指导功能基因组学研究。
计算工作流自动化
LLM智能体可以自动化复杂的计算工作流。例如,scChat能够通过自然语言交互,对单细胞和空间转录组学数据进行端到端解释。CellAgent则能自主构建自适应的多组学分析流程。Cell-Designer甚至能根据高层次任务描述,直接从原始组学数据构建细胞行为的预测模型。
全栈研究
全栈研究智能体旨在自动化从问题提出到科学发现的整个科研流程。BioSmart能在通用计算生物学环境中自主分析多样的组学数据,产生新见解。CellOracle则通过迭代提出基因扰动、模拟结果、评估并修正假设的闭环,专注于功能基因组学和疾病机制发现。
优化
为了提升LLM智能体的可靠性和准确性,引入了多种优化策略。
- 后期训练:通过强化学习对模型进行后期训练,可以显著提升其在生物医学任务中的表现。
- 自验证:智能体在推理时通过实时交叉引用权威生物数据库来验证其输出,从而减少幻觉。
- 自进化:一些智能体系统能够随时间积累知识并改进其推理策略。例如,Evolving-Bio-Agent通过双循环系统实现自进化:一个用于任务执行的“反思-再规划”循环,另一个用于在人类专家反馈下演进推理模板库。
结论与未来工作
本文全面综述了LLM在虚拟细胞研究中的应用,并提出了一个分为“预言机”和“智能体”两大范式的方法分类体系。尽管取得了显著进展,但未来仍面临重要的挑战和机遇:
- 可扩展性(Scalability)
- LLM预言机:需要将分子级和组学级的多模态序列统一到连贯的联合表示中,并采用能处理超长细胞上下文的高效架构。
- LLM智能体:关键在于开发长时记忆机制,以在冗长的实验工作流中维持连贯的推理和上下文感知。
- 泛化性(Generalizability)
- LLM预言机:对未见过细胞类型的泛化仍是巨大挑战,需要更严格且具生物学意义的基准测试。
- LLM智能体:目前缺乏系统和公平的评估框架,这阻碍了对其优缺点的深入理解。
- 可靠性(Reliability)
- LLM预言机:需要确保模拟的稳定性和可复现性,并通过不确定性估计和可解释性来量化预测置信度。
- LLM智能体:需要行为一致性,并利用不确定性和可解释性使其决策过程可被理解和验证。
局限性
本综述主要关注LLMs与虚拟细胞研究的交叉领域。细胞成像研究本身是一个广阔而丰富的领域,但鉴于其范围巨大,本文并未广泛覆盖。未来的工作可能会扩大范围,以提供对这些领域更全面的审视。